合作研究发现希舞症致病基因新功能—CDKL5通过相分离介导突触后可塑性
CDKL5 Deficiency Disorder(CDD,也称”希舞症”),是一种X染色体基因突变引发的罕见神经发育疾病。CDKL5(Cyclin-dependent kinase-like 5)基因位于X染色体,其突变会导致严重的神经系统功能损伤,以难治性癫痫与智力发育障碍为核心症状。目前,该突变尚无有效的治疗手段,给患者及家庭带来了深重的痛苦与负担。2026年2月19日,中国科学院脑科学与智能技术卓越创新中心(神经科学研究所)、上海脑科学与类脑研究中心熊志奇团队、上海交通大学BIO-X研究院朱金伟团队以及上海交通大学医学院附属精神卫生中心朱永川团队,合作研究揭示了CDKL5蛋白具备相分离性质,CDKL5借由该性质参与神经细胞的突触后可塑性,而基因突变导致的兴奋性突触后分子相分离受损,是希舞症学习记忆损伤表现的潜在机制,论文发表在PNAS期刊上。
在缺失CDKL5蛋白的小鼠模型中,可观察到包含多种学习记忆功能受损在内的疾病表型,并伴随着突触形态和功能可塑性异常。在细胞层次,突触为承担学习与记忆功能的最小元件,而树突棘为特化的兴奋性突触后结构,其响应神经活动而发生的形态及功能的改变,构成神经可塑性的基础。然而在分子层面,可塑性所依赖的突触后重塑的过程及分子机制尚不完全明确。前期研究表明CDKL5蛋白富集于突触后且与以PSD95为代表的突触后蛋白互作,加之CDKL5缺乏症的学习记忆功能受损表型,提示CDKL5可能参与突触可塑性,但目前仍然缺乏直接的证据及具体分子机制。
近年来,生物大分子液液相分离(liquid-liquid phase separation, LLPS)现象被广泛发现参与各种细胞的生理过程,液液相分离的典型表现为生物大分子自发凝聚形成液态的凝聚体,此过程由多价且较弱的分子间相互作用驱动。树突棘的突触后致密带(postsynaptic density, PSD)的组装同样依赖于蛋白质的液液相分离,而CDKL5的相互作用蛋白PSD95为突触后致密带凝聚体组装的核心脚手架蛋白之一。
本研究结合生物物理与神经生物学的多层次实验手段,揭示CDKL5在突触后的分子生化机制。首先,在蛋白质层面CDKL5蛋白具有独立相分离的能力。虽然激酶区有所贡献,但CDKL5的相分离主要依赖于羟基端的内在无序区。同时,CDKL5的羟基末端还具有一个可以和PSD95的PDZ结构域发生多价相互作用的PDZ结合基序,介导CDKL5与PSD95的共同相分离来促进CDKL5定位至突触后致密带。细胞层次上,在离体培养的原代神经元中敲低CDKL5会导致树突棘形态变得细长,对应较弱的树突棘功能强度,且CDKL5敲低会阻止树突棘响应长时程增强刺激导致的增大,提示CDKL5贡献于正常树突棘形态。CDKL5可响应神经元活动进入或离开树突棘,突变CDKL5使其相分离缺陷可阻止CDKL5的动态运输。在受到化学诱导的长时程抑制刺激后CDKL5离开树突棘,并伴随树突棘和突触后致密带的缩小。反之,受到化学诱导的长时程增强刺激后CDKL5富集至树突棘,通过与PSD95的共同相分离促进PSD增大。在富集至树突棘过程中CDKL5还借助自身相分离性质招募Kalirin7蛋白来激活Rac1信号通路,促进细胞骨架的组装与分支,使之与突触后致密带的扩大相协同,共同作用于树突棘总体形态的改变,表明CDKL5贡献于树突棘形态的可塑性。脑片电生理记录显示回补野生型的CDKL5蛋白可挽救CDKL5蛋白缺失导致的突触后长时程增强受损,而回补相分离功能缺失的CDKL5突变型蛋白无法挽救该缺陷,进一步表明突触后功能的可塑性依赖于CDKL5的相分离。最后,临床相关错义突变可影响CDKL5凝聚体的物理性质,并损伤CDKL5的突触后定位,动态运输,对Kalirin7的招募,及PSD的尺寸和树突棘密度。
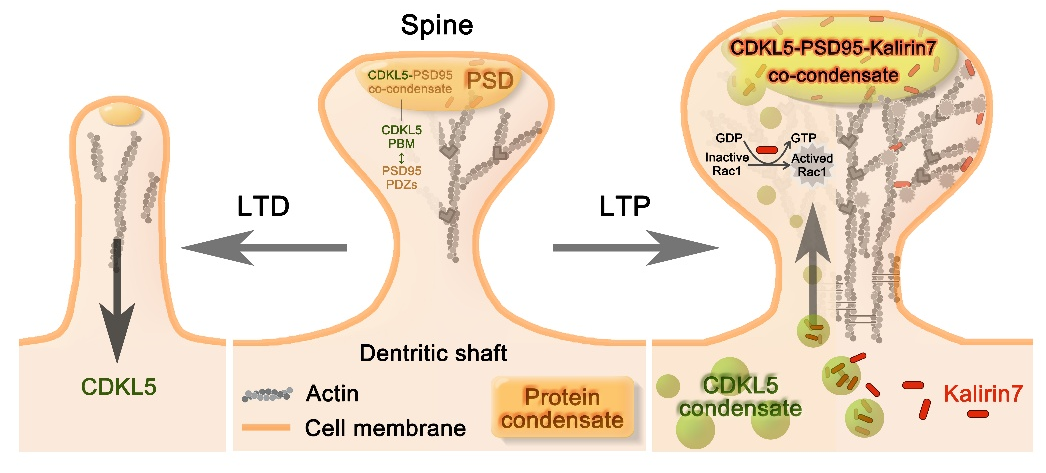
CDKL5相分离在突触后可塑性发生过程中的功能示意图
CDKL5通过与PSD95的共同相分离定位于突触后(中),长时程抑制刺激导致CDKL5离开突触后并伴随树突棘的缩小(左),长时程增强刺激后CDKL5通过相分离携带Kalirin7进入突触后,协调PSD扩大与actin的分支与组装,共同导致树突棘的增大(右)。
综上,CDKL5的相分离参与树突棘的形态及功能的可塑性,而CDKL5相分离的损伤为CDKL5缺乏症的病理机制之一。此研究不仅解析了CDKL5蛋白的全新性质与功能,揭示了CDKL5缺乏症学习记忆功能损伤相关症状的直接分子机制,也增进了对于相分离现象如何参与神经元生理功能,及相分离损伤致病机制的理解。